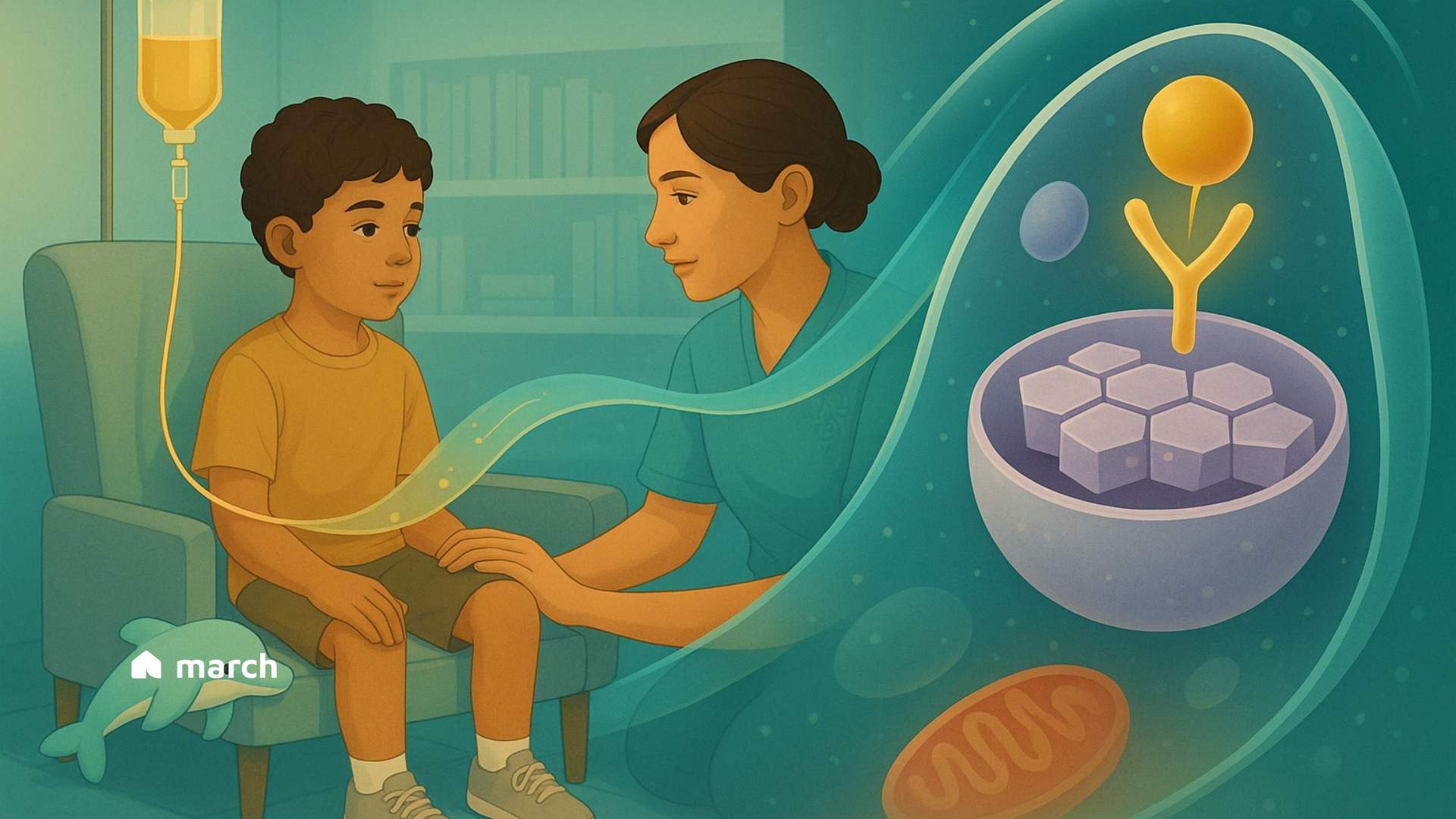
The journey that follows the diagnosis of a rare condition, like a Lysosomal Storage Disease (LSD), often begins with a deep desire for understanding. These conditions, of which there are over 70, start on a microscopic level (Platt et al., 2018). They are inherited metabolic disorders that result from a defect in a single gene (Meikle et al., 1999). This defect leads to the deficiency or absence of a specific enzyme within the lysosomes of our cells (Platt et al., 2018). Lysosomes function as the cell’s recycling center, breaking down complex molecules like fats and sugars into simpler components for the cell to reuse (Parenti et al., 2015). When a specific lysosomal enzyme is missing or faulty, its target substance accumulates, causing progressive damage to cells, tissues, and organs (Platt et al., 2018). This can lead to a wide range of symptoms, including developmental delay, seizures, enlarged organs, and bone abnormalities (National Institute of Neurological Disorders and Stroke, 2024). Finding clarity in the science can be a grounding first step, helping to chart the path forward with more confidence.
For many living with an LSD, a remarkable therapy has changed the future of what’s possible. Enzyme Replacement Therapy (ERT) works by directly supplying the missing or faulty enzyme the body needs, getting to the very heart of the issue (Beck, 2018). For many, it has meant a significant reduction in symptoms, an improvement in quality of life, and a renewed sense of hope in managing a chronic condition (Giugliani et al., 2018).
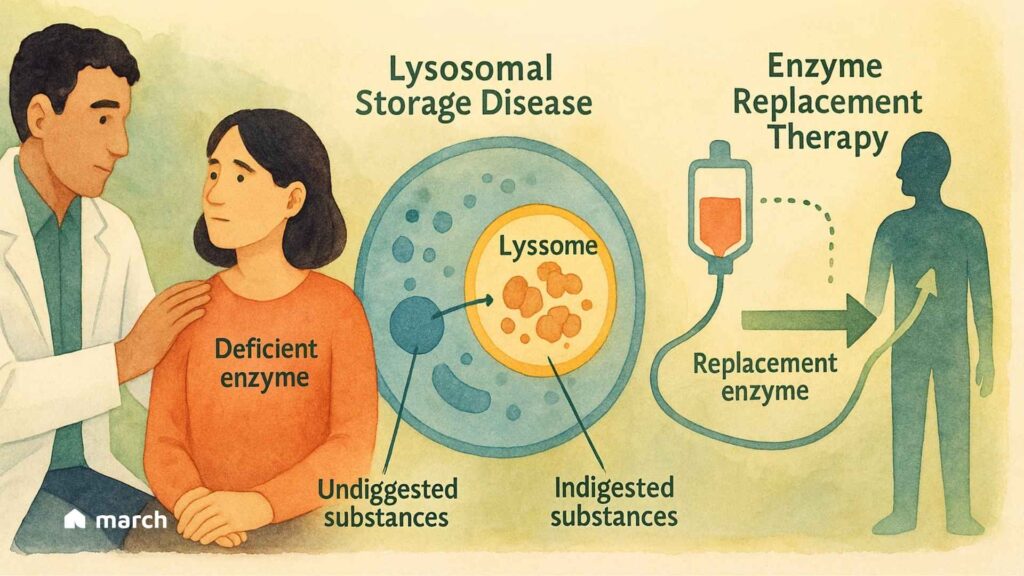
Think of the missing enzyme as a specialized worker who is absent from their post. ERT is designed to deliver a functional version of this exact worker directly to the cells that need it. Given through an intravenous (IV) infusion, the therapy travels through the bloodstream (Aflaki et al., 2018; Beck, 2018). The replacement enzymes are engineered with a special “address label,” often a mannose-6-phosphate sugar molecule, that the body’s cells can recognize via specific receptors (Kornfeld, 1986; Park & Lee, 2022). This allows the cells to welcome the new enzyme inside and guide it directly to the lysosomes where the buildup is happening (Kornfeld, 1986). Once there, it can get to work, helping to break down the stored materials and restore a healthier balance within the cell (Park & Lee, 2022).
It is important to understand that ERT is a lifelong treatment, not a cure, as it does not correct the underlying genetic mutation (Platt et al., 2018; Beck, 2018). The therapy has limitations, as the large enzyme molecules cannot easily cross the blood-brain barrier, making it less effective for neurological symptoms (Begley, 2015; Pardridge, 2015). Despite these challenges, understanding the science behind new therapies can be incredibly empowering. It helps foster more informed conversations and a greater sense of control on a journey that can often feel overwhelming.
To explore this topic on an even deeper level and immerse yourself in the fascinating story of ERT, we encourage you to listen to our podcast episode. It delves into the intricate science behind Enzyme Replacement Therapy.
References:
Aflaki, E., et al. (2018). Enzyme replacement therapies: what is the best option?. Current Pharmaceutical Design, 24(11), 1238-1250.
Beck, M. (2018). Enzyme replacement therapy and beyond—in memoriam Roscoe O. Brady, M.D. (1923–2016). Journal of Inherited Metabolic Disease, 41(1), 3-13.
Begley, D. J. (2015). Modifying blood–brain barrier transport to bring hope for patients with lysosomal storage diseases. Journal of Cerebral Blood Flow & Metabolism, 35(1), 3-5.
Giugliani, R., et al. (2018). Enzyme replacement therapy for mucopolysaccharidoses: new developments and clinical outcomes. Expert Opinion on Orphan Drugs, 6(4), 277-287.
Kornfeld, S. (1986). Trafficking of lysosomal enzymes in mammalian cells. FASEB Journal, 1(6), 462-468.
Meikle, P. J., et al. (1999). Prevalence of lysosomal storage disorders in Australia. JAMA, 281(3), 249-254.
National Institute of Neurological Disorders and Stroke. (2024). Lysosomal Storage Diseases Fact Sheet. National Institutes of Health.
Pardridge, W. M. (2015). Blood–Brain Barrier Targeting of Therapeutic Lysosomal Enzymes. In Lysosomal Storage Disorders (pp. 327-343). Springer.
Parenti, G., Andria, G., & Ballabio, A. (2015). Lysosomal Storage Diseases: From Pathophysiology to Therapy. In The Metabolic & Molecular Bases of Inherited Disease (Chapter 131). McGraw-Hill Education.
Park, J. J., & Lee, K. (2022). Mannose-6-phosphate glycan for lysosomal targeting: various applications from enzyme replacement therapy to lysosome-targeting chimeras. Animal Cells and Systems, 26(3), 84-91.
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F., & Tifft, C. J. (2018). Lysosomal storage diseases. Nature Reviews Disease Primers, 4(1), 27.